‘Attacks of ventricular fibrillation following exertion or emotional disturbance, a prolonged QT interval on cardiogram and a familial incidence’.1 The now pathognomonic trio of symptoms and signs of the Long QT Syndrome (LQTS) as described by Professor Ward in his 1964 Journal of the Irish Medical Association case series publication. A 6-year-old girl, suffering from repeated syncope, had been referred for cardiology review by her tenacious GP who thus far, had unsuccessfully consulted widely on this troublesome and novel case. The child was admitted to hospital and her symptoms recreated by running her around the ward – she collapsed, pulseless and unconscious. The electrocardiographic changes are punctiliously described – marked QT prolongation at baseline and ‘bizarre’ ventricular extrasystoles degenerating into ventricular fibrillation of an ‘abnormal configuration’1. We now know this to represent Torsades de pointes.
It was noted that clinical examination and basic biological testing were normal. Based on the astute observations of the author, it was correctly concluded that this new disorder was an abnormality of repolarisation – ‘as evidenced by the normal interval between the first and second heart sounds, the abnormality is confined to the recovery phase in which the heart prepares for the next contraction’1. Furthermore, it was suggested that ‘Undue sensitivity of the myocardium to sympathetic stimulation’1 may underlie the condition, given attacks occurred during stress and the dilated pupils of the child. In terms of heritability, it was noted that the child’s brother was also affected. Another brother had a normal electrocardiogram (ECG), as did their father however, the mother’s ECG showed QT prolongation. It was concluded from these observations that this cardiac syndrome had an autosomal dominant inheritance pattern2.
Fortuitously, the IMJ article was picked up by The Lancet and published as an Annotation in their July issue 19643. They noted that it was the ‘first time this condition had been described’ and they recommended that an ECG be carried out in fainting children. Perhaps even more fortuitously, the annotation was noticed by Caesaro Romano, a Genoese paediatrician4 who had also recently published a series of three siblings suffering from syncopal events, abnormal QT interval and T waves and ventricular fibrillation on cardiogram5. Following reports of further cases from groups in Sweden6 and South Africa7, publication of the eleventh case in 1970 referred to the condition as the Romano-Ward Syndrome8, the term that was widely adopted thereafter. Of note, the Professors Romano and Ward never actually met each other.9
Both Ward and Romano commented on the similarities between their cases and the previously described Jervell Lange-Nielsen syndrome (JLN). In 1957, two Norwegian physicians published10 the ‘obscure’ case of 4 siblings (from a family of 6) each suffering from deaf mutism (sic) and fainting attacks. Consistent with the cases reported by Romano and Ward, the attacks happened soon after effort, the ECG was normal apart from a prolonged QT interval and clinical examination was unremarkable. The children all died suddenly. As none of the children in Romano or Ward’s series were deaf, it was considered that the conditions were linked but separate entities. With remarkable foresight however, Ward noted that the cardio-auditory JLN syndrome was likely to represent the autosomal recessive form of the Romano-Ward Syndrome.2
Meanwhile, in the North of Ireland, at Queen’s University Belfast, Professor (now Sir) Peter Froggatt was collaborating with researchers in Oxford and Detroit11 to perform ECGs on congenitally deaf people throughout Ireland and Britain. This was a very large endeavour at the time (1964) and they succeeded in assessing the QT interval of 1460 patients. From these, nine new cases of JLN were identified, four of them from Ireland. In order to define the normal limits of the QT interval in their population, they conducted ECGs in a control group of 369 Belfast school children and measured the distribution of the QT interval. The group also produced a regression equation to normalise QT for age, sex and heart rate. This equation was essentially a paediatric QT correction formula.11 In their discussion, the authors touch on some of the issues that remain pertinent in the Long QT community today. They note that, even within families, penetrance (as measured by the QT interval) seems to be varied and not necessarily linked to fatality. Even 50 years later in the era of genetic testing, while risk stratification in LQTS is somewhat more accurate, the same questions in regard to variable presentation of LQTS genotypes remain.12
A decade later, in one of his first publications on the subject (now totalling >160), Peter Schwartz reported 6 new cases of long QT in both deaf and normal hearing children.13 This brought the total reported cases to 203. This particular publication is notable for several reasons. First, it described the successful therapeutic use of beta-blockers, and based on an earlier case report from Arthur Moss14, they also performed a left stellate ganglion sympathectomy, which successfully shortened the QT interval and rendered the patient syncope-free. Second, this paper also introduced the key concept of the importance of T wave morphology in addition to the QT interval, made the distinction between acquired and congenital LQTS and again commented on the unanswered questions regarding risk stratification and penetrance. Additionally, the umbrella term ‘Long QT Syndrome’, encompassing both Romano-Ward and Jervell Lange-Nielsen, first appeared in this article.
Two pivotal publications on LQTS appeared in 1985. Schwartz formulated diagnostic criteria, particularly useful in borderline cases15 and the first report from the International Long QT registry was published in Circulation16. The follow-up data on 146 patients provided significant insight into the natural history of the syndrome and identified risk factors for sudden death – namely congenital deafness, history of syncope, female gender and documented Torsade de pointes or ventricular fibrillation. They noted that the absolute QT interval was not necessarily proportional to mortality risk and indeed, that the QT interval was actually normal (<440ms) in several patients with documented syncope, Torsade de pointes and family history. This was an important observation, as we now know that there is considerable overlap in the QTc intervals of controls versus LQTS mutation carriers.17
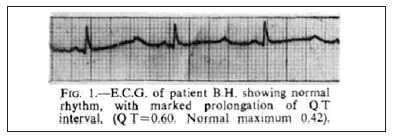
The early to mid nineties saw the most fundamental advances in the unravelling of the pathogenesis of LQTS. Genetic linkage studies performed by Mark Keating’s group (Utah) in 1991 mapped a gene locus on Chromosome 11.18 In 1994, they mapped further loci on chromosomes 3 and 7.19 The following year, in a seminal single issue of Cell, mutations in the genes KCNH2/hERG (encoding a voltage-gated potassium channel)20 and SCN5A (encoding a voltage-gated sodium channel),21 were identified as the cause of LQT subclass 2 and subclass 3 respectively. By 1996, with the discovery of potassium channel KVLQT1as the LQT 1 linked gene (KCNQ1)22 a molecular basis for the majority of congenital LQT syndrome cases had been established. With a growing awareness of the Long QT Syndrome amongst cardiologists, updated and more refined diagnostic criteria were required. Schwartz’s new points-based criteria recognised the spectrum of disease (graded scale based on the QTc value), sex differences in QT interval and the importance of T wave morphology.23 These diagnostic criteria remain in use today. In relation to T wave morphology, we note that in Figure 1 of Ward’s original article1 (Figure 1), subtle T wave notching is evident in the first beat of the ECG. This suggests that the patient was suffering from LQT subclass 2.
Figure 1: The original article from OC Ward, Journal of the Irish Medical Association 1964. Note the subtle T wave notching of the first beat of the ECG
2005 marked the 25th anniversary of the International Long QT registry.24 The well-defined and phenotyped clinical pedigrees now contained in the registry (families from both North America and Europe) provided not just insights to the clinical natural history of the LQT spectrum but also the biochemical material for genetic analyses. The ever-present transatlantic cooperation in the Long QT syndrome was further evident in 2013 with the publication of a joint Heart Rhythm Society / European Heart Rhythm Association statement on the Diagnosis and Management of Patients with inherited Primary Arrhythmia syndromes.25 This most recent guidance endorsed the LQTS risk score model, the therapeutic use of beta-blockers with implantable cardiac defibrillators in selected cases and the use of specific genetic testing. Despite the extraordinary progress in the characterisation, pathophysiology and treatment of the Long QT syndrome over the short period of fifty years, questions still remain. Risk stratification remains imprecise. The advances in the molecular aspects of the disease have enabled mutation-specific risk assessment26 but it has also been demonstrated that the presence or absence of QT-modifying single nucleotide polymorphisms act as a ‘second hit’ to the mutation and either prolong or indeed shorten the QT interval.27 Of particular clinical importance, is the need to improve identification of the highest risk patients who would benefit from implantation of a cardiac defibrillator.
Over the past fifty years, advances in the field of inherited cardiac arrhythmias have been rapid and substantial. However, as Ward demonstrated 1964, precise and accurate description of the phenotype remains key when dealing with novel diseases.
Correspondence: JI Vandenberg
Mark Cowley Lidwill Research Programme in Cardiac Electrophysiology, Molecular Cardiology and Biophysics division, Victor Chang Cardiac Research Institute, Darlinghurst, NSW 2010, Australia
Email: [email protected]
References
1. Ward OC. A new familial cardiac syndrome in children. J. Ir. Med. Assoc.1964:54:103-6
2. Ward OC. The electrocardiographic abnormality in familial cardiac arrhythmia. Ir J Med Sci 1966;41:553–557
3. Annotation. Congenital cardiac arrhythmia. Lancet 1964;2:26-27?14.
4. Romano C. Congenital cardiac arrhythmia. Letter. Lancet.1965;1:658-659 Letter?15.
5. Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’eta’ pediatrica. Clinica Pediatrica 1963;45:656-83
6. Gamstorp I, Nilsen R, Westling H. Congenital Cardiac Arrhythmia. Lancet.1964 Oct 31;2:965.
7. Barlow JB, Bosman CK, Cochrane JWC.Congenital cardiac arrhythmia. Letter. Lancet. 1964;2:531
8. Karhunen P, Luomanmäki K, Heikkilä J, Eisalo A. Syncope and Q-Tprolongation without deafness: The Romano-Ward syndrome. American Heart Journal, 1970; 80: 820-3.
9. Ward C. Long QT syndromes: the Irish dimension. Ir Med J 2005;98: 120–2.
10. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the QT interval, and sudden death. American Heart Journal, 1957; 54:59–68.
11. Fraser GR, Froggatt P, James TN. Congenital deafness associated with electrocardiographic abnormalities, fainting attacks and sudden death. A recessive syndrome. Q J Med. 1964; 33:361–85.
12. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini MV, Nastoli J, Bottelli G, Folli R, Cappelletti D.. Risk stratification in the Long-QT Syndrome. N Engl J Med. 2003 May 8;348:1866–74.
13. Schwartz PJ, Periti M, Malliani A. The Long QT Syndrome. American Heart Journal 1975;89:378–90.
14. Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for thetreatment of long QT interval syndrome. N Engl J Med. 1971 Oct14;285:903–4.
15. Schwartz PJ. Idiopathic long QT syndrome: progress and questions. American Heart Journal 1985;109:399–411.
16. Moss AJ, Schwartz PJ, Crampton RS, Locati E, Carleen E. The long QT syndrome: a prospective international study. Circ 1985;71:17–21.
17. Viskin S. The QT interval: Too long, too short or just right. Heart Rhythm. 2009May;6:711–5.
18. Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science,1991; 252:704–6.
19. Jiang C, Atkinson D, Towbin JA, Splawski I, Lehmann MH, Li H, Timothy K, Taggart T, Schwartz PJ, Vincent GM, Moss AJ, Keating MT. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994 Oct;8:141–7.
20. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell.1995 80, 795–803.
21. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell1995;80:805-11
22. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, deJager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996 Jan;12:17–23.
23. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circ. 1993 Aug 1;88:782–4.
24. Moss AJ. 25th Anniversary of the International Long-QT Syndrome Registry: An Ongoing Quest to Uncover the Secrets of Long-QT Syndrome. Circ 2005;111:1199–1201
25. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Burgada J, Chiang C-E, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PF, Shimizu W, Tomaselli G, Tracy C . HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm 2013;10:1932–63
26. Migdalovich D, Moss AJ, Lopes CM, Costa J, Ouellet G, Barsheshet A, McNitt S, Polonsky S, Robinson JL, Kaufman ES, Platonov PG, Shimizu W, Towbin JA, Vincent GM, Wilde AAM, Goldenberg I. Mutation and gender-specific risk in type 2long QT syndrome: implications for risk stratification for life-threatening cardiac events in patients with long QT syndrome. Heart Rhythm, 2011;8:1537–43.
27. Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, Spooner PM, Priori SG.. Polymorphisms in the NOS1AP Gene Modulate QT Interval Duration and Risk of Arrhythmias in the Long QT Syndrome. JACC. 2010 Jun;55:2745–52.