|
|
|
|
|
|
|
|
Anna Molloy,Orla Cotter,Brian Sweeney
|
|
|
|
Ir Med J. 2012 Jun;105(6):186-7
A Molloy, O Cotter, R van Spaendonk, E Sistermans, B Sweeney
Department of Neurology, Cork University Hospital, Wilton, Cork
Abstract
The hereditary leukodystrophies are rare disorders caused by molecular abnormalities leading to destruction of or failure of development of central white matter. For almost 30 years there has been increasing recognition of later onset Autosomal Dominant Leukodystrophy (ADLD). We report the first genetically confirmed case of lamin B1 duplication causing ADLD from Ireland.
|
|
Case Report
A 47 year- old gentleman presented with a six- year history of urinary dysfunction including hesitancy, frequency and nocturia. He described increasing fatigue and hearing loss more marked on the left side over the preceding 18 months. In his past history he had had a severe upper gastrointestinal bleed secondary to duodenal ulcer with a marked hypotensive episode peri- operatively. Of note in his family history, his mother had died of a progressive neurological illness at age 57. This had been labelled as a ‘brain tumour’ but no confirmatory evidence had been provided. Of three male siblings, two had died of myocardial infarction in their forties. Another had a diagnosis of bipolar affective disorder.
On initial examination, there were no cognitive deficits noted. He had an ataxic gait, Romberg’s sign was positive. Cranial nerves were intact. Tone and power were normal on limb examination. Reflexes were 2+ throughout and plantar responses were flexor. There was past-pointing suggesting cerebellar dysfunction bilaterally. Blood results were within normal limits or negative including: vitamin B12, folate, thyroid function tests, cortisol levels, very long chain fatty acids, HIV, HTLV-1, Lymedisease antibodies, ACE. Genetic testing for CADASIL was negative. Cerebrospinal Fluid analysis revealed normal cell count, cytology, protein, glucose with negative oligoclonal bands. An audiogram revealed asymmetric bilateral sensorineural hearing loss. Visual evoked responses showed dispersed low amplitude responses, consistent with possible optic nerve axonal loss. Electroretinogram showed mild delay bilaterally but acceptable amplitudes were noted, implying possible optic nerve pathology.
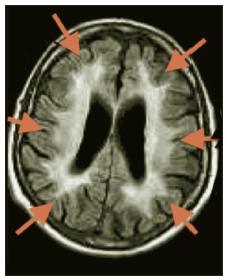
MRI brain scan showed widespread white matter disease in centrum semiovale and both cerebral and cerebellar peduncles without enhancement. The corpus callosum was spared. Long tract involvement in midbrain, pons and medulla was noted. Over the succeeding 6 years he continued to progress with symptoms including increasing fatigue, ataxia and falls, dysarthria, and ongoing urinary frequency and urgency. Progression of neurological signs was noted including ataxia, dysarthria, spasticity in all 4 limbs, and bilateral extensor plantars. MRI showed deterioration with extensive signal change in the centrum semiovale, pyramidal tracts, midbrain, cerebellar peduncles, medial lemnisci and signal changes from medulla to cervical cord. Ventricles were dilated in keeping with a degree of atrophy (Figures 1 and 2). Because of the clinical and MRI findings, LMNB1 (Lamin B1) testing was performed. A positive result for LMNB1 exon 1 t/m 11 duplication was obtained consistent with a diagnosis of ADLD. MLPA analysis was used to detect the duplication, using the P071 kit from MRC-Holland (more details are available on mlpa.com).
Figure 1: Involvement of cerebellar peduncles on FLAIR MRI scan.
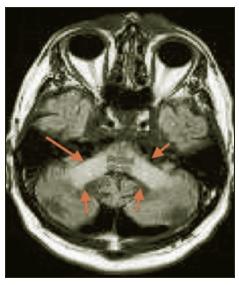
Figure 2: Diffuse predominantly fronto- parietal white matter changes on T2W imaging in advanced stages.
Discussion
This patient had an 8-10 year history of progressive multi-system neurological dysfunction with MRI showing a marked diffuse leukodystrophy. There was a family history of a progressive early onset neurological disorder in his mother which now raises the possibility of her being affected by ADLD. The diagnosis carries significant implications for this patient’s sibling and children, as it has been highly penetrant in the families described so far. ADLD due to Lamin B1 duplication was first described in 1984 by Eldridge et al in an Irish-American kindred.1 Symptom onset was in the fourth or fifth decade with autonomic dysfunction preceeding motor impairment and ataxia as in our patient. Slow progression of symptoms to death after 20 years was typical. MRI characteristics in kindreds described include diffuse white matter changes in a symmetrical distribution bilaterally, most prominent in fronto-parietal areas and cerebellar peduncles. Spinal cord involvement has been noted, as has cerebral, cerebellar and medullary atrophy.2 Sparing of optic radiations initially is characteristic, unlike many other leukodystrophies. In our case the imaging findings were typical.
ADLD is the first human disease known to be caused by a mutation of the gene responsible for lamin B1, and is the only laminopathy caused by over expression.3 Excess folding and blebbing of the nuclear envelope has been shown to occur at a cellular level, and the extent of expression is crucial in regulation of oligodendrocyte maturation and myelin formation.4 Worldwide at this point, nine families have been described, including two of Irish- American origin.5,6 Brussino et al reported an Italian kindred with a form of ADLD without LMNB1 duplication, the autonomic features were not present at the outset, raising the possibility that other genetic causes for variant forms of ADLD exist.7 We are unaware of any connection between previous cases and our patient at this time.
Correspondence: A Molloy
Department of Neurology, St Vincent’s University Hospital, Elm Park, Dublin 4
Email: [email protected]
Acknowledgements
We would like to thank Professor Marjo van der Knapp and Dr Erik Sistermans for their help in the diagnosis of this case.
References
1. Eldridge R, Anayiotos CP, Schlesinger S, Cowen D, Bever C, Patronas N, Mc Farland H. Hereditary adult onset leukodystrophy simulating chronic progressive multiple sclerosis. N Eng J Med 1984;311:948- 953.
2. Schwankhaus JD, Patronas N, Dorwart R, Eldridge R, Schlesinger S, McFarland H. Computed tomography and magnetic resonance imaging in adult- onset leukodystrophy. Arch Neurol 1988;45:1004-1008.
3. Padiath Q, Saigoh K, Schiffmann R, Ashara H, Yamada T, Koeppen A, Hogan K, Ptácek LJ, Fu YH.Lamin B1 duplications cause autosomal dominant leukodystrophy. Nature Gen 2006;10:1115- 1123.
4. Lin ST, Fu YH. Mir-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis Mod Mech 2009. 2:178-188.
5. Coffeen CM, McKenna CE, Koeppen AH, Plaster NM, Maragakis N, Mihalopoulos J, Schwankhaus JD, Flanigan KM, Gregg RG, Ptácek LJ, Fu YH.Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Mol Genet 2000;9:787- 793.
6. Padiath QS,Fu YH. Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol 2010;98:337-357.
7. Brussino A, Vaula G, Cagnoli C, Panza E, Seri M, Di Gregorio E, Scappaticci S, Camanini S, Daniele D, Bradac GB, Pinessi L, Cavalieri S, Grosso E, Migone N, Brusco A. A family with autosomal dominant leukodystrophy linked to lamin B1 mutations. Eur J Neurol 2010;17:541-549.
|
|
|
|
Author's Correspondence
|
|
No Author Comments
|
|
|
Acknowledgement
|
|
No Acknowledgement
|
|
|
Other References
|
|
No Other References
|
|
|
|
|