|
|
|
|
|
|
|
|
M Iqbal,S Connolly,Yvonne Langan,J Redmond
|
|
|
Ir Med J. 2012 Jun;105(6):183-4
M Iqbal, S Connolly, Y Langan, J Redmond
Department of Neurology, St James’s Hospital, James’s St, Dublin 8
Abstract
Lambert-Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder of neuromuscular transmission (NMJ) that shares many clinical features with myasthenia gravis (MG). We report a 73 year-old lady who presented 10 years previously with stiffness of both calves, dry mouth, fatigue, proximal weakness and areflexia in lower limbs. Neurophysiological studies were consistent with LEMS. Her work up for an underlying neoplasm was negative. She recently developed unilateral ptosis and diplopia which dramatically improved with pyridostigmine suggesting concomitant MG.
|
|
Introduction
Lambert-Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder of neuromuscular transmission (NMJ) that shares many clinical features with myasthenia gravis (MG). Although there has been controversy about the coexistence of the two syndromes in the same patient, several case reports in the past 30 years have described this rare association1.
Case Report
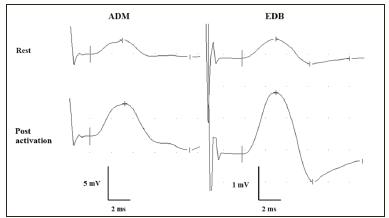
A 73 year-old lady presented 10 years previously with stiffness of both calves, dry mouth and fatigue over a 6-month period. Initial examination revealed proximal muscle weakness and areflexia in lower limbs. Routine investigations were unremarkable. Nerve conduction studies revealed reduced compound muscle action potential amplitudes at rest in upper and lower limbs with potentiation >100% following voluntary activation for 10 seconds (see Figure). Significant decrement was also elicited by repetitive nerve supramaximal stimulation at 3/second. Single fibre electromyographic recordings made from tibialis anterior muscle revealed markedly increased jitter and blocking. The findings were consistent with a presynaptic neuromuscular transmission disorder.
Investigations for an underlying neoplasm including bronchoscopy, computed tomogram (CT) thorax and VGCC-ab were unremarkable. The diagnosis was deemed to be LEMS of probable autoimmune aetiology. She had mild response to low-dose corticosteroids, azathioprine and 3,4 diaminopyridine. A trial of intravenous immunoglobulin (IVIg) over the next few years conferred no sustained clinical benefit. After 8 years, she received two courses of plasma exchange therapy that resulted in full resolution of symptoms. During an admission for a second course of plasma exchange treatment, she developed fatigable left eye ptosis and diplopia without pupillary abnormality. Urgent magnetic resonance imaging (MRI) and magnetic resonance angiogram (MRA) brain were unremarkable. Neurophysiological testing revealed partially treated LEMS and was not diagnostic of MG.
However, a post synaptic disorder was suspected on clinical grounds and she was given a trial of pyridostigmine from which she had an immediate dramatic clinical response. She had no peripheral features of MG. It was concluded that she had concomitant MG with LEMS. Subsequent AchR-ab, antibodies to muscle specific tyrosine kinase (MuSK-ab) and other autoimmune screen were negative. She did not receive any further plasma exchange therapy and her symptoms recurred after 6 months. Ocular symptoms also recurred when pyridostigmine was withdrawn.
Figure 1: Compound muscle action potentials recorded from abductor digiti minimi (ADM) and extensor digitorum brevis (EDB) muscles after supramaximal stimulation of ulnar and peroneal nerve respectively, at rest and following maximal voluntary activation for 10 seconds. Post activation potentiation was 166% (ADM) and 257 % (EDB).
Discussion
Because of their overlapping clinical symptoms, many cases of LEMS may be misdiagnosed as MG. Patients with LEMS tend to have more lower-extremity weakness, and MG patients have more oculobulbar symptoms, although there is considerable overlap. In a review of 50 LEMS patients all patients had proximal leg weakness, but 25 patients had diplopia, 21 had ptosis, 12 had dysarthria, 12 had dysphagia, and 8 had difficulty in chewing2. Oculobulbar symptoms were found in 78% of 23 LEMS patients at the Lahey Clinic3. Oculobulbar symptoms were rare in 38 LEMS patients in another study and these symptoms were only counted if there was no limb involvement4. In MG, compound muscle action potential (CMAP) amplitudes are rarely reduced, and decrement occurs at low and high rates of stimulation5. In LEMS, CMAP amplitudes are reduced; decrement occurs at low rates of stimulation, but at high rates a several-fold increment occurs6. Generally, serum P/Q VGCC-ab can be detected in 85% of LEMS patients7. AChR-ab are present in approximately 80% of MG patients and a variable percentage of the remaining patients have MuSK-ab8,9. Some investigators have suggested that the rare finding of AChR-ab in LEMS patients is “a non-pathogenic epiphenomenon” of no clinical significance10.
In our patient the clinical features and neurophysiological studies initially confirmed the diagnosis of LEMS. However, the occurrence of fatigable ptosis and extraocular findings 10 years later in our patient was unusual and is not well described in LEMS. There is no plausible explanation for the dramatic clinical response to pyridostigmine in our patient with these findings other than associated MG with LEMS.
Correspondence: M Iqbal
Department of Neurology, St James’s Hospital, James’s St, Dublin 8
Email: [email protected]
References
1. Newsom-Davis J, Leys K, Vincent A, Ferguson I, Modi G, Mills K. Immunological evidence for the co-existence of the Lambert-Eaton myasthenic syndrome and myasthenia gravis in two patients. J Neurol Neurosurg Psychiatry. 1991;54:452-453.
2. O'Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain : a journal of neurology 1988;111:577-596.
3. Burns TM, Russell JA, LaChance DH, Jones HR. Oculobulbar involvement is typical with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53:270-273.
4. Wirtz PW, Sotodeh M, Nijnuis M, Van Doorn PA, Van Engelen BG, Hintzen RQ, De Kort PL, Kuks JB, Twijnstra A, De Visser M, Visser LH, Wokke JH, Wintzen AR, Verschuuren JJ. Difference in distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73:766-768.
5. Mayer RF, Williams IR. Incrementing responses in myasthenia gravis. Arch Neurol 1974;31:24-26.
6. Lambert EH, Rooke ED. Myasthenic state and lung cancer. In: Brain RW, Norris FH, eds. The remote effects of cancer on the nervous system. New York: Grune & Stratton, 1965: 67-80.
7. Martin-Moutot N, Haro L, Santos RG, Mori Y, Seagar M. Phoneutria nigriventer omega-Phonetoxin IIA: a new tool for anti-calcium channel autoantibody assays in Lambert-Eaton myasthenic syndrome. Neurobiol Dis. 2006;22:57-63.
8. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365-368.
9. Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve. 1992;15:720-724.
10. Katz JS, Wolfe GI, Bryan WW, Tintner R, Barohn RJ. Acetylcholine receptor antibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1998;50:470-475.
|
|
|
|
Author's Correspondence
|
|
No Author Comments
|
|
|
Acknowledgement
|
|
No Acknowledgement
|
|
|
Other References
|
|
No Other References
|
|
|
|
|